
![]()
Tidak mampu beli software Turbomole, Gaussian, HyperChem dan sejenisnya adalah takdir kita sekarang. Semoga suatu saat dimampukan! Sementara berpuas diri dengan software bajakan atau memilih software kimia komputasi open source. Salah satunya adalah ERGOSCF.
Ergo adalah program kimia kuantum (kimia komputasi) untuk hitungan SCF skala besar.
Fitur kuncinya:
- Melakukan hitungan struktur elektronik menggunakan Hartree-Fock and Kohn-Sham density functional theory.
- Ditulis dalam C++.
- Menggunakan basis sets Gaussian. Cek di https://bse.pnl.gov/bse/
- Elektron inti dan valensi dimasukkan dalam hitungan.
- Model restricted and unrestricted diimplementasikan untuk hitungan energi.
- Mengimplementasikan jangkauan luas, baik pure dan hybrid Kohn-Sham density functionals.
- Menggunakan teknik modern linear scaling, seperti fast multipole methods, hierarchic sparse matrix algebra, density matrix purification, dan efficient integral screening.
- Linear scaling dicapai tidak hanya untuk CPU usage tetapi juga memory utilization.
- Konsumsi waktu bagian kode program secara paralel menggunakan paradiga shared-memory.
Download:
1) http://ftp.debian.org/debian/pool/main/e/ergo/ergo_3.4.0.orig.tar.gz
2) http://ergoscf.org/source/tarfiles/ergo-3.4.tar.gz
3) http://ergoscf.org/source.php
Instalasi:
ErgoSCF sudah ada dalam repositori Debian, sehingga untuk instalasinya sangat mudah, cukup ketik di terminal:
sudo apt-get install ergo
maka ErgoSCF akan terpasang di sistem linux kita. Versi Ergo terakhir yang ada di repositori Debian adalah versi 3.3.1. Sedangkan versi terbaru sudah versi 3.4. Untuk instalasi versi 3.4 silahkan merujuk ke link tutorialnya di http://ergoscf.org/tutorial.php.
Basis Set:
Ergo versi 3.3.1 yang dipasang dari repositori Debian akan tersimpan di sistem linux pada direktori /usr/bin, sedangkan basis set-nya tersimpan di direktori /usr/share/ergo/basis. Pada versi 3.3.1 ada 111 basis set, dalam versi 3.4 ada 113. basis set, lihat pada gambar berikut:
Karena menggunakan basis set Gaussian, tentunya jumlah basis set dapat ditambahkan sendiri sesuai keperluan. Cek di https://bse.pnl.gov/bse/
Running Ergo:
Cara 1:
1. Buatlah file sh menggunakan notepad atau editor lainnya, misal menggunakan gedit.
2. Dalam editor ketikkan spesifikasi molekulnya, perhatikan contoh berikut untuk molekul air :
ergo <<EOINPUT
molecule_inline
O 0.0 0.0 0.0
H -1.809 0.0 0.0
H 0.453549 1.751221 0.0
EOF
basis = "6-31Gss"
run "HF"
EOINPUT
Keterangan:
1) ergo adalah command untuk memanggil dan menjalankan ergo.
2) Di antara EOINPUT berisi spesifikasi molekul, jenis basis set yang digunakan dan metode kalkulasi yang dipakai
3) Spesifikasi molekul diakhiri dengan EOF = End of File.
3. Simpanlah, misal dengan nama h2o.sh
4. Sebaiknya setiap file input itu disimpan dalam folder tersendiri.
5. Jalankan ergo untuk menggunakan file input itu dengan cara, ketik di terminal: sh h2o.sh, dan tunggu sampai selesai. Jangan lupa menjalankan terminalnya di folder tempat menyimpan file h2o.sh.
6. Jika hitungan sudah selesai maka akan dihasilkan 2 file, file pertama ergoscf.out dan file kedua density.bin. File out adalah file teks output hasil hitungan, dapat dibuka dengan editor, sedangkan file bin dapat digunakan lebih lanjut untuk proses lain.
7. Sebaiknya segera ganti nama file ergoscf.out dengkan nama lain supaya tidak tertumpuk dengan hasil hitungan berikutnya.
Cara 2:
1. Ketikkan langsung teks sh pada cara 1 di terminal, BARIS DEMI BARIS.
Di baris 1, ketik: ergo <<EOINPUT [Enter]
Di baris 2, ketik: molecule_inline [Enter]
Di baris 3, ketik: O 0.0 0.0 0.0 [Enter]
Di baris 4, ketik: H -1.809 0.0 0.0 [Enter]
Di baris 5, ketik: H 0.453549 1.751221 0.0 [Enter]
Di baris 6, ketik: EOF [Enter]
Di baris 7, ketik: basis = "6-31Gss" [Enter]
Di baris 8, ketik: run "HF" [Enter]
Di baris 9, ketik: EOINPUT [Enter]
2. Enter terakhir di atas otomatis akan menjalankan ergo, tunggu sampai selesai. Hasilnya adalah 2 file, 1 file out dan 1 file bin. Lihat gambar di bawah ini.
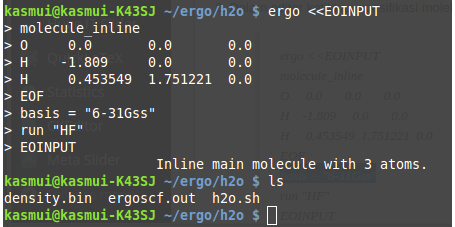
file h2o.sh pada gambar di atas adalah file hasil cara 1.
3. File out dapat dilihat dengan menggunakan editor seperti cara 1.
Cara 3:
1. Cara ini memanggil file spesifikasi molekul yang sudah ada, seperti ekstensi xyz dan mol
2. Misal kita buat dulu file mol-nya menggunakan editor sebagai berikut:
BASIS
6-31Gss
Direct SCF calculation w/o symmetry
———————————–
2 0 X Y
8. 1
O 0.0 0.0 0.0
1. 2
H -1.809 0.0 0.0
H 0.453549 1.751221 0.0
3. Simpan dengan nama h2o.mol
4. Masuk ke folder tempat menyimpan file h2o.mol.
5. Masuk terminal dan ketikkan sebagai berikut:
ergo <<EOINPUT
molecule "h2o.mol"
run "HF"
EOINPUT
Setiap baris di aras harus diakhiri dengan [Enter] untuk menuju baris berikutnya.
6. [Enter] terakhir akan langsung menjalankan ergo dengan input h2o.mol di atas.
7. Tunggu sampai selesai, hasilnya akan sama dengan cara 1 dan 2.
8. Cara 3 ini dapat juga dilakukan menggunakan cara pertama.
CATATAN:
1) Untuk hitungan yang lebih lanjut, silahkan merujuk langsung ke tutorialnya di http://ergoscf.org/tutorial.php.
2) Jika bingung bagaimana menjalankan, ketik saja di terminal: ergo -h, untuk meminta bantuan.
Referensi: http://ergoscf.org/